Our Research
As a large comprehensive cohort of Canadian patients with pulmonary fibrosis, CARE-PF provides a network for future clinical research and a robust platform for translational and basic science research that is unequalled in Canada and among the leaders globally. CARE-PF has supported over 40 publications on topics that are directly relevant to the care of people living with pulmonary fibrosis. These include studies on the diagnosis, management, and outcomes of pulmonary fibrosis, which have been conducted with collaborators from around the world.
Our Publications
The Canadian Registry for Pulmonary Fibrosis: Design and Rationale of a National Pulmonary Fibrosis Registry
Abstract
Background: The relative rarity and diversity of fibrotic interstitial lung disease (ILD) have made it challenging to study these diseases in single-centre cohorts. Here we describe formation of a multicentre Canadian registry that is needed to describe the outcomes of fibrotic ILD and to enable detailed healthcare utilization analyses that will be the cornerstone for future healthcare planning.
Methods: The Canadian Registry for Pulmonary Fibrosis (CARE-PF) is a prospective cohort anticipated to consist of at least 2,800 patients with fibrotic ILD. CARE-PF will be used to (1) describe the natural history of fibrotic ILD, specifically determining the incidence and outcomes of acute exacerbations of ILD subtypes and (2) determine the impact of ILD and acute exacerbations of ILD on health services use and healthcare costs in the Canadian population. Consecutive patients with fibrotic ILD will be recruited from five Canadian ILD centres over a period of five years. Patients will be followed up as clinically indicated and will complete standardized questionnaires at each clinic visit. Prespecified outcomes and health services use will be measured based on self-report and linkage to provincial health administrative databases.
Conclusion: CARE-PF will be among the largest prospective multicentre ILD registries in the world, providing detailed data on the natural history of fibrotic ILD and the healthcare resources used by these patients. As the largest and most comprehensive cohort of Canadian ILD patients, CARE-PF establishes a network for future clinical research and early phase clinical trials and provides a platform for translational and basic science research.
Baseline characteristics and comorbidities in the CAnadian REgistry for Pulmonary Fibrosis
Abstract
Background: The CAnadian REgistry for Pulmonary Fibrosis (CARE-PF) is a multi-center, prospective registry designed to study the natural history of fibrotic interstitial lung disease (ILD) in adults. The aim of this cross-sectional sub-study was to describe the baseline characteristics, risk factors, and comorbidities of patients enrolled in CARE-PF to date.
Methods: Patients completed study questionnaires and clinical measurements at enrollment and each follow-up visit. Environmental exposures were assessed by patient self-report and comorbidities by the Charlson Comorbidity Index (CCI). Baseline characteristics, exposures, and comorbidities were described for the overall study population and for incident cases, and were compared across ILD subtypes.
Results: The full cohort included 1285 patients with ILD (961 incident cases (74.8%)). Diagnoses included connective tissue disease-associated ILD (33.3%), idiopathic pulmonary fibrosis (IPF) (24.7%), unclassifiable ILD (22.3%), chronic hypersensitivity pneumonitis (HP) (7.5%), sarcoidosis (3.2%), non-IPF idiopathic interstitial pneumonias (3.0%, including idiopathic nonspecific interstitial pneumonia (NSIP) in 0.9%), and other ILDs (6.0%). Patient-reported exposures were most frequent amongst chronic HP, but common across all ILD subtypes. The CCI was ≤2 in 81% of patients, with a narrow distribution and range of values.
Conclusions: CTD-ILD, IPF, and unclassifiable ILD made up 80% of ILD diagnoses at ILD referral centers in Canada, while idiopathic NSIP was rare when adhering to recommended diagnostic criteria. CCI had a very narrow distribution across our cohort suggesting it may be a poor discriminator in assessing the impact of comorbidities on patients with ILD.
Inhalational exposures in patients with fibrotic interstitial lung disease: Presentation, pulmonary function and survival in the Canadian Registry for Pulmonary Fibrosis
Abstract
Background and objective: Inhalational exposures are a known cause of interstitial lung disease (ILD), but little is understood about their prevalence across ILD subtypes and their relationship with pulmonary function and survival.
Methods: Patients with fibrotic ILD were identified from the multicentre Canadian Registry for Pulmonary Fibrosis. Patients completed questionnaires regarding ILD-related occupational and environmental exposures. The relationship between exposures and the outcomes of baseline age, gender, family history, pulmonary function and survival was analysed using linear and logistic regression models, linear mixed-effect regression models and survival analysis using multivariable Cox proportional hazards along with the log-rank test.
Results: There were 3820 patients included in this study, with 2385 (62%) having ILD-related inhalational exposure. Exposed patients were younger, particularly in the idiopathic pulmonary fibrosis subgroup. Inhalational exposure was associated with male gender (adjusted OR 1.46, 95% CI 1.28-1.68, p < 0.001) and family history of pulmonary fibrosis (adjusted OR 1.73, 95% CI 1.40-2.15, p < 0.001). Patients with any inhalational exposure had improved transplant-free survival (hazard ratio 0.81, 95% CI 0.71-0.92, p = 0.001); this effect persisted across diagnostic subtypes. The relationship between exposures and annual change in forced vital capacity varied by ILD subtype.
Conclusion: Patients with fibrotic ILD report high prevalence of inhalational exposures across ILD subtypes. These exposures were associated with younger age at diagnosis, male gender and family history of pulmonary fibrosis. Identification of an inhalational exposure was associated with a survival benefit. These findings suggest that inhaled exposures may impact clinical outcomes in patients with ILD, and future work should characterize the mechanisms underlying these relationships.
Keywords: CARE-PF; Canadian Registry for Pulmonary Fibrosis; fibrotic interstitial lung disease; inhalational exposure; occupational exposure.
Characteristics of pulse oximetry and arterial blood gas in patients with fibrotic interstitial lung disease
Abstract
Background: Fibrotic interstitial lung disease (ILD) is frequently associated with abnormal oxygenation; however, little is known about the accuracy of oxygen saturation by pulse oximetry (SpO2) compared with arterial blood gas (ABG) saturation (SaO2), the factors that influence the partial pressure of carbon dioxide (PaCO2) and the impact of PaCO2 on outcomes in patients with fibrotic ILD.
Study design and methods: Patients with fibrotic ILD enrolled in a large prospective registry with a room air ABG were included. Prespecified analyses included testing the correlation between SaO2 and SpO2, the difference between SaO2 and SpO2, the association of baseline characteristics with both the difference between SaO2 and SpO2 and the PaCO2, the association of baseline characteristics with acid-base category, and the association of PaCO2 and acid-base category with time to death or transplant.
Results: A total of 532 patients with fibrotic ILD were included. Mean resting SaO2 was 92±4% and SpO2 was 95±3%. Mean PaCO2 was 38±6 mmHg, with 135 patients having PaCO2 <35 mmHg and 62 having PaCO2 >45 mmHg. Correlation between SaO2 and SpO2 was mild to moderate (r=0.39), with SpO2 on average 3.0% higher than SaO2. No baseline characteristics were associated with the difference in SaO2 and SpO2. Variables associated with either elevated or abnormal (elevated or low) PaCO2 included higher smoking pack-years and lower baseline forced vital capacity (FVC). Lower baseline lung function was associated with an increased risk of chronic respiratory acidosis. PaCO2 and acid-base status were not associated with time to death or transplant.
Interpretation: SaO2 and SpO2 are weakly-to-moderately correlated in fibrotic ILD, with limited ability to accurately predict this difference. Abnormal PaCO2 was associated with baseline FVC but was not associated with outcomes.
Keywords: interstitial fibrosis
Association of BMI and Change in Weight With Mortality in Patients With Fibrotic Interstitial Lung Disease
Abstract
Background: Mortality risk assessment in interstitial lung disease (ILD) is challenging. Our objective was to determine the prognostic significance of BMI and change in weight in the most common fibrotic ILD subtypes.
Research question: Could BMI and weight loss over time be reliable prognostic indicators in patients with fibrotic ILD?
Study design and methods: This observational retrospective multicenter cohort study enrolled patients with fibrotic ILD from the six-center CAnadian REgistry for Pulmonary Fibrosis (CARE-PF, derivation) and the ILD registry at the University of California, San Francisco (UCSF, validation). Patients were subcategorized as underweight (BMI < 18.5), normal weight (BMI 18.5-24.9), overweight (BMI 25-29.9), or obese (BMI > 30). Annual change in weight was calculated for all years of follow-up as the slope of best fit using the least square method based on every available measurement. Separate multivariable analyses evaluated the associations of BMI and change in weight with mortality, adjusting for common prognostic variables.
Results: The derivation and validation cohorts included 1,786 and 1,779 patients, respectively. Compared with patients with normal BMI, mortality was highest in patients who were underweight (hazard ratio [HR], 3.19; 95% CI, 1.88-5.43; P < .001) and was lowest in those who were overweight (HR, 0.52; 95% CI, 0.36-0.75; P < .001) or obese (HR, 0.55; 95%CI, 0.37-0.83; P < .001) in the analysis adjusted for the ILD-GAP (gender, age, physiology) Index. Patients who had a weight loss of at least 2 kg within 1 year had increased risk of death in the subsequent year (HR, 1.41; 95% CI, 1.01-1.97; P = .04) after adjustment for the ILD-GAP Index and baseline BMI category, with a plateau in risk for patients with greater weight loss. Consistent results were observed in the validation cohort.
Interpretation: Both BMI and weight loss are independently associated with 1-year mortality in fibrotic ILD. BMI and weight loss may be clinically useful prognostic indicators in fibrotic ILD.
Keywords: BMI; change in weight; idiopathic pulmonary fibrosis; interstitial lung disease; prognosis.
Travel Distance to Subspecialty Clinic and Outcomes in Patients with Fibrotic Interstitial Lung Disease
Abstract
Rationale: Early access to subspecialty care is associated with improved outcomes for patients with fibrotic interstitial lung disease (ILD). Access to ILD care may be limited for patients living far from subspecialty clinics.
Objectives: To test the hypothesis that greater travel distance to access ILD clinical care would be associated with more severe disease at time of referral and worse clinical outcomes.
Methods: Patients with fibrotic ILD were recruited from a multicenter national pulmonary fibrosis registry. Residential postal codes were geocoded to estimate travel distance from the home to the clinic. Travel distance was dichotomized at ⩽70 km (near) and >70 km (far). Demographics and disease severity at the initial referral, changes in lung function, and the risk of death or lung transplant were analyzed in unadjusted and adjusted models for their association with travel distance.
Results: The cohort included 1,162 patients, of whom 856 lived near to their ILD clinic and 306 lived far from their ILD clinic. Patients residing farther from their clinic were younger, more likely to have smoked, had a greater 6-minute-walk distance, and had lower composite risk scores than patients residing closer to their clinic. In models adjusted for age, sex, and baseline forced vital capacity, patients from farther away had a greater risk of death or lung transplant than patients residing closer (hazard ratio, 1.52; 95% confidence interval [CI], 1.10–2.11), a finding predominantly driven by patients with connective tissue disease–related ILD (hazard ratio, 2.14; 95% CI, 1.16–3.94).
Conclusions: Patients with fibrotic ILD with a longer travel distance to their ILD clinic had better prognostic indices at baseline but had a higher risk of death or lung transplant in the total cohort and in patients with connective tissue disease–related ILD. Assuming that disease epidemiology and severity are distributed evenly across geographic regions, these findings raise important questions about equitable access to patient care in large healthcare regions with centralized subspecialty programs.
Keywords: pulmonary fibrosis; epidemiology; registries; geography
Costs of Workplace Productivity Loss in Patients With Fibrotic Interstitial Lung Disease
Abstract
Background: Fibrotic interstitial lung diseases (ILDs) are highly morbid chronic disorders that frequently occur in working age individuals. The goal of this study was to determine workplace productivity loss, its determinants, and its estimated costs in patients with fibrotic ILD.
Methods: Patients with idiopathic pulmonary fibrosis, chronic hypersensitivity pneumonitis, idiopathic nonspecific interstitial pneumonia, or unclassifiable ILD were identified from the six-center Canadian Registry for Pulmonary Fibrosis (CARE-PF). The Work Productivity and Activity Impairment questionnaire was used to determine health-related productivity loss. Independent predictors of low workplace productivity were identified by using multivariate regression. Patient data were compared with Canadian population census data. The average productivity loss (hours per week) and the individual’s hourly wage were used to estimate the costs of productivity loss.
Results: Of 650 eligible patients, 148 (23%) were employed. Productivity loss was reported by 55% of employed patients with an average productivity loss of 7.8 ± 0.9 h per week (2.3 ± 0.6 h per week related to absenteeism and 5.5 ± 0.6 h per week related to presenteeism). Employment among patients with ILD aged 25 to 54 years was 23% lower than the age- and sex-matched general Canadian population (60% vs 83%; P < .001). Employment among patients with ILD aged ≥ 55 years was 18% lower than in the age- and sex-matched population (20% vs 38%; P < .001). Dyspnea and cough were independent predictors of workplace productivity loss. Estimated annual costs of productivity loss were 11,610 Canadian dollars per employee with ILD.
Conclusions: Workplace productivity loss is common in fibrotic ILD, strongly correlated with symptom severity, and associated with significant cost.
Keywords: economics; hypersensitivity pneumonia; idiopathic pulmonary fibrosis; interstitial lung disease.
Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry
Abstract
Background: Progressive fibrosing interstitial lung disease (PF-ILD) is characterised by progressive physiological, symptomatic and/or radiographic worsening. The real-world prevalence and characteristics of PF-ILD remain uncertain.
Methods: Patients were enrolled from the Canadian Registry for Pulmonary Fibrosis between 2015 and 2020. PF-ILD was defined as a relative forced vital capacity (FVC) decline ≥10%, death, lung transplantation or any two of: relative FVC decline ≥5% and <10%, worsening respiratory symptoms or worsening fibrosis on computed tomography of the chest, all within 24 months of diagnosis. Time-to-event analysis compared progression between key diagnostic subgroups. Characteristics associated with progression were determined by multivariable regression.
Results: Of 2746 patients with fibrotic ILD (mean±sd age 65±12 years; 51% female), 1376 (50%) met PF-ILD criteria in the first 24 months of follow-up. PF-ILD occurred in 427 (59%) patients with idiopathic pulmonary fibrosis (IPF), 125 (58%) with fibrotic hypersensitivity pneumonitis (HP), 281 (51%) with unclassifiable ILD (U-ILD) and 402 (45%) with connective tissue disease-associated ILD (CTD-ILD). Compared with IPF, time to progression was similar in patients with HP (hazard ratio (HR) 0.96, 95% CI 0.79-1.17), but was delayed in patients with U-ILD (HR 0.82, 95% CI 0.71-0.96) and CTD-ILD (HR 0.65, 95% CI 0.56-0.74). Background treatment varied across diagnostic subtypes, with 66% of IPF patients receiving antifibrotic therapy, while immunomodulatory therapy was utilised in 49%, 61% and 37% of patients with CHP, CTD-ILD and U-ILD, respectively. Increasing age, male sex, gastro-oesophageal reflux disease and lower baseline pulmonary function were independently associated with progression.
Conclusions: Progression is common in patients with fibrotic ILD, and is similarly prevalent in HP and IPF. Routinely collected variables help identify patients at risk for progression and may guide therapeutic strategies.
Association of Particulate Matter Exposure With Lung Function and Mortality Among Patients With Fibrotic Interstitial Lung Disease
Abstract
Importance: Particulate matter 2.5 μm or less in diameter (PM2.5) is associated with adverse outcomes for patients with idiopathic pulmonary fibrosis, but its association with other fibrotic interstitial lung diseases (fILDs) and the association of PM2.5 composition with adverse outcomes remain unclear.
Objective: To investigate the association of PM2.5 exposure with mortality and lung function among patients with fILD.
Design, Setting, and Participants: In this multicenter, international, prospective cohort study, patients were enrolled in the Simmons Center for Interstitial Lung Disease Registry at the University of Pittsburgh in Pittsburgh, Pennsylvania; 42 sites of the Pulmonary Fibrosis Foundation Registry; and 8 sites of the Canadian Registry for Pulmonary Fibrosis. A total of 6683 patients with fILD were included (Simmons, 1424; Pulmonary Fibrosis Foundation, 1870; and Canadian Registry for Pulmonary Fibrosis, 3389). Data were analyzed from June 1, 2021, to August 2, 2022.
Exposures: Exposure to PM2.5 and its constituents was estimated with hybrid models, combining satellite-derived aerosol optical depth with chemical transport models and ground-based PM2.5 measurements.
Main Outcomes and Measures: Multivariable linear regression was used to test associations of exposures 5 years before enrollment with baseline forced vital capacity and diffusion capacity for carbon monoxide. Multivariable Cox models were used to test associations of exposure in the 5 years before censoring with mortality, and linear mixed models were used to test associations of exposure with a decrease in lung function. Multiconstituent analyses were performed with quantile-based g-computation. Cohort effect estimates were meta-analyzed. Models were adjusted for age, sex, smoking history, race, a socioeconomic variable, and site (only for Pulmonary Fibrosis Foundation and Canadian Registry for Pulmonary Fibrosis cohorts).
Results: Median follow-up across the 3 cohorts was 2.9 years (IQR, 1.5-4.5 years), with death for 28% of patients and lung transplant for 10% of patients. Of the 6683 patients in the cohort, 3653 were men (55%), 205 were Black (3.1%), and 5609 were White (84.0%). Median (IQR) age at enrollment across all cohorts was 66 (58-73) years. A PM2.5 exposure of 8 μg/m3 or more was associated with a hazard ratio for mortality of 4.40 (95% CI, 3.51-5.51) in the Simmons cohort, 1.71 (95% CI, 1.32-2.21) in the Pulmonary Fibrosis Foundation cohort, and 1.45 (95% CI, 1.18-1.79) in the Canadian Registry for Pulmonary Fibrosis cohort. Increasing exposure to sulfate, nitrate, and ammonium PM2.5 constituents was associated with increased mortality across all cohorts, and multiconstituent models demonstrated that these constituents tended to be associated with the most adverse outcomes with regard to mortality and baseline lung function. Meta-analyses revealed consistent associations of exposure to sulfate and ammonium with mortality and with the rate of decrease in forced vital capacity and diffusion capacity of carbon monoxide and an association of increasing levels of PM2.5 multiconstituent mixture with all outcomes.
Conclusions and Relevance: This cohort study found that exposure to PM2.5 was associated with baseline severity, disease progression, and mortality among patients with fILD and that sulfate, ammonium, and nitrate constituents were associated with the most harm, highlighting the need for reductions in human-derived sources of pollution.
Integration and Application of Radiologic Patterns From Clinical Practice Guidelines on Idiopathic Pulmonary Fibrosis and Fibrotic Hypersensitivity Pneumonitis
Abstract
Background: Clinical practice guidelines separately describe radiologic patterns of usual interstitial pneumonia (UIP) and fibrotic hypersensitivity pneumonitis (fHP), without direction on whether or how to apply these approaches concurrently within a single patient.
Research question: How can we integrate guideline-defined radiologic patterns to diagnose interstitial lung disease (ILD) and what are the pitfalls associated with described patterns that require reassessment in future guidelines?
Study design and methods: Patients from the Canadian Registry for Pulmonary Fibrosis underwent detailed reevaluation in standardized multidisciplinary discussion. CT scan features were quantified by chest radiologists masked to clinical data, and guideline-defined patterns were assigned. Clinical data then were provided to the radiologist and an ILD clinician, who jointly determined the leading diagnosis.
Results: Clinical-radiologic diagnosis in 1,593 patients was idiopathic pulmonary fibrosis (IPF) in 26%, fHP in 12%, connective tissue disease-associated ILD (CTD-ILD) in 34%, idiopathic pneumonia with autoimmune features in 12%, and unclassifiable ILD in 10%. Typical and probable UIP patterns corresponded to a diagnosis of IPF in 66% and 57% of patients, respectively. Typical fHP pattern corresponded to an fHP clinical diagnosis in 65% of patients, whereas compatible fHP was nonspecific and associated with CTD-ILD or IPAF in 48% of patients. No pattern ruled out CTD-ILD. Gas trapping affecting > 5% of lung parenchyma on expiratory imaging was an important feature broadly separating compatible and typical fHP from other patterns (sensitivity, 0.77; specificity, 0.91).
Interpretation: An integrated approach to guideline-defined UIP and fHP patterns is feasible and supports > 5% gas trapping as an important branch point. Typical or probable UIP and typical fHP patterns have moderate predictive values for a corresponding diagnosis of IPF and fHP, although occasionally confounded by CTD-ILD; compatible fHP is nonspecific.
Keywords: hypersensitivity pneumonitis; idiopathic pulmonary fibrosis; interstitial lung disease; multidisciplinary discussion; usual interstitial pneumonia.
Impact of surgical lung biopsy on lung function and survival in patients with idiopathic pulmonary fibrosis in a multi-centre registry cohort
Abstract
Background and ojective: Establishing an accurate and timely diagnosis of idiopathic pulmonary fibrosis (IPF) is essential for appropriate management and prognostication. In some cases, surgical lung biopsy (SLB) is performed but carries non-negligible risk. The objective of this retrospective study was to determine if SLB is associated with accelerated lung function decline in patients with IPF using the Canadian Registry for Pulmonary Fibrosis.
Methods: Linear mixed models and Cox proportional hazards regression models were used to compare decline in forced vital capacity (FVC)%, diffusion capacity of the lung (DLCO%) and risk of death or lung transplantation between SLB and non-SLB patients. Adjustments were made for baseline age, sex, smoking history, antifibrotic use, and lung function. A similar analysis compared lung function changes 12 months pre- and post-SLB.
Results: A total of 81 SLB patients and 468 non-SLB patients were included. In the SLB group, the post-biopsy annual FVC% decline was 2.0% (±0.8) in unadjusted, and 2.1% (±0.8) in adjusted models. There was no difference in FVC% decline, DLCO% decline, or time to death or lung transplantation between the two groups, in adjusted or unadjusted models (all p-values >0.07). In the pre-post SLB group, no differences were identified in FVC% decline in unadjusted or adjusted models (p = 0.07 for both).
Conclusion: No association between SLB and lung function decline or risk of death or lung transplantation was identified in this multi-centre study of patients with IPF.
Keywords: idiopathic pulmonary fibrosis; interstitial lung disease; lung function; lung transplantation; outcomes; risk of death; surgical lung biopsy.
Neighborhood-Level Disadvantage Impacts on Patients with Fibrotic Interstitial Lung Disease
Abstract
Rationale: Fibrotic interstitial lung disease (fILD) is a group of pathologic entities characterized by scarring of the lungs and high morbidity and mortality. Research investigating how socioeconomic and residential factors impact outcomes in patients with fILD is lacking.
Objectives: To determine the association between neighborhood-level disadvantage and presentation severity, disease progression, lung transplantation, and mortality in patients with fILD from the United States and Canada.
Methods: We performed a multicenter, international, prospective cohort study of 4,729 patients with fILD from one U.S. and eight Canadian ILD registry sites. Neighborhood-level disadvantage was measured by the area deprivation index in the United States and the Canadian Index of Multiple Deprivation in Canada.
Measurements and Main Results: In the U.S. but not in the Canadian cohort, patients with fILD living in neighborhoods with the greatest disadvantage (top quartile) experience the highest risk of mortality (hazard ratio = 1.51, P = 0.002), and in subgroups of patients with idiopathic pulmonary fibrosis, the top quartile of disadvantage experienced the lowest odds of lung transplantation (odds ratio = 0.46, P = 0.04). Greater disadvantage was associated with reduced baseline DLCO in both cohorts, but it was not associated with baseline FVC or FVC or DLCO decline in either cohort.
Conclusions: Patients with fILD who live in areas with greater neighborhood-level disadvantage in the United States experience higher mortality, and patients with idiopathic pulmonary fibrosis experience lower odds of lung transplantation. These disparities are not seen in Canadian patients, which may indicate differences in access to care between the United States and Canada.
Keywords: health equity; healthcare disparities; interstitial lung disease; residence characteristics; social determinants of health.
Treatment Initiation in Patients with Interstitial Lung Disease in Canada
Abstract
Rationale: Real-life pharmacological treatment patterns of patients with interstitial lung diseases (ILD) remain elusive.
Objectives: To determine how often and with what medications patients with ILD are treated in Canadian tertiary care clinics.
Methods: All patients with ILD prospectively enrolled in the Canadian Registry for Pulmonary Fibrosis were included in this observational study. All first instances of medication for each patient were compiled. The time between the diagnosis of ILD and the first initiation of an ILD-related medication was compared across diagnostic categories. Cox proportional hazards models were used to identify variables associated with time-to-treatment initiation, stratified by diagnostic category.
Results: Out of 2,652 patients, a total of 1,483 (56%) were treated with an ILD-related medication during the median follow up of 3.0 years (1.4-5.9), including 349/646 (54%) patients with idiopathic pulmonary fibrosis (IPF) who received an antifibrotic. Patients with IPF were treated earlier and in greater proportion than those with non-IPF ILD (P = 0.001). Male sex and lower lung function were associated with shorter time-to-treatment initiation in the full cohort.
Conclusions: Overall, 56% of patients with ILD seen across seven Canadian specialized ILD clinics received pharmacological treatment over a median follow up of 3 years. Further studies are needed to assess longitudinal patterns of treatment and their influence on key outcomes.
Keywords: idiopathic pulmonary fibrosis; interstitial; lung diseases; therapeutics.
Mapping EQ5D utilities from forced vital capacity and diffusing capacity in fibrotic interstitial lung disease
Abstract
Objectives: Fibrotic interstitial lung disease (ILD) includes a large group of conditions that lead to scarring of the lungs. The lack of available 5-level EuroQol 5D (EQ5D) data has limited the ability to conduct economic evaluations in ILD. The purpose of this study was to develop and validate a mapping algorithm that predicts EQ5D utilities from commonly collected pulmonary function measurements (forced vital capacity [FVC] and diffusing capacity of the lung for carbon monoxide [DLCO]) in fibrotic ILDs.
Methods: EQ5D utility and pulmonary function measurements from the Canadian Registry for Pulmonary Fibrosis were included. Ordinary least squares (OLS), beta regression, two-part, and tobit models were used to map EQ5D utilities from FVC or DLCO. Model performance was assessed by comparing the predicted and observed utilities. Subgroup analyses were also conducted to test how well models performed across different patient characteristics. The models were then externally validated in the Australian Idiopathic Pulmonary Fibrosis Registry.
Results: The OLS model performed as well as other more complex models (root mean squared error: 0.17 for FVC and 0.16 for DLCO). As with the other models, the OLS algorithm performed well across the different subgroups (except for EQ5D utilities < 0.5) and in the external validation cohort.
Conclusion: We developed a mapping algorithm that predicts EQ5D utilities from FVC and DLCO, with the intent that this algorithm can be applied to clinical trial populations and real-world cohorts that have not prioritized collection of health-related utilities. The mapping algorithm can be used in future economic evaluations of potential ILD therapies.
Association of BMI with pulmonary function, functional capacity, symptoms, and quality of life in ILD
Abstract
Obesity is a health epidemic associated with greater morbidity and mortality in the general population. Mass loading of the thorax from obesity leads to a restrictive pulmonary defect that reduces lung capacity in obese individuals without pulmonary disease, and may exacerbate the restrictive pulmonary physiology that is characteristic of interstitial lung disease (ILD). The purpose of this study was to test the association of body mass index (BMI) with pulmonary function, functional capacity, and patient-reported outcomes (dyspnea and quality of life) in patients with ILD. We analyzed 3169 patients with fibrotic ILD from the Canadian Registry for Pulmonary Fibrosis. Patients were subcategorized as underweight (BMI<18.5 kg/m2), normal weight (18.5≤BMI<25), overweight (25≤BMI<30), obese I (30≤BMI<35), obese II (35≤BMI<40), and obese III (BMI>40). Analysis was performed using a linear regression with adjustment for common prognostic variables. Overweight and obese BMI categories were associated with worse pulmonary function, functional capacity, dyspnea, and quality of life compared to normal weight. This is likely a result of mass loading on the thorax, and we speculate that intentional weight-loss may improve lung function and functional capacity in obese patients with fibrotic ILD. The underweight BMI category was also associated with worse functional capacity compared to normal weight, which may reflect greater disease severity or the presence of other comorbidities. Future work should explore the clinical utility of BMI to improve patient outcomes.
Keywords: Body mass index; Interstitial lung disease; Morbidity; Pulmonary function.
Sex-and Race-Based Differences in the Treatment of Interstitial Lung Diseases in North America and Australasia
Abstract
Background: Biological sex, gender, and race are important considerations in patients with interstitial lung diseases (ILDs).
Research question: Does a patient’s sex assigned at birth, and race, influence ILD treatment initiation?
Study design and methods: Patients with ILD from three longitudinal prospective registries were compared in this observational study. ILD-related medications included antifibrotics and immunomodulating medications. Race was dichotomized as “White” vs “non-White.” Time to treatment initiation was determined from the date of the initial ILD registry visit to the date of first medication initiation. Proportions of treated patients were compared between groups by χ2 test. Cox proportional analysis was used to determine how sex and race were associated with time to treatment initiation stratified by ILD diagnosis.
Results: A total of 4,572 patients were included across all cohorts. The proportion of men who received treatment was higher than for women in the Canadian cohort (47% vs 40%; P < .001), and the proportion of White patients who received treatment was also higher compared with non-White patients (46% vs 36%; P < .001). In contrast, the proportion of treated men in the Chicago cohort was lower compared with women (56% vs 64%; P = .005), and that of White patients was lower compared with non-White patients (56% vs 69%; P < .001). No sex- or race-based differences in proportions of patients treated were found in the Australasian cohort. White race was significantly associated with earlier treatment initiation compared with non-White race across diagnoses in the Canadian cohort, whereas the opposite association was found in the Australasian cohort.
Interpretation: Sex- and race-based differences exist in the initiation of ILD treatment, with variability across different cohorts in different countries. Reasons for these differences need to be further explored in future studies.
Keywords: idiopathic pulmonary fibrosis; interstitial lung disease; race; sex and gender; treatment.
Effect of continued antifibrotic therapy after forced vital capacity decline in patients with idiopathic pulmonary fibrosis; a real world multicenter cohort study
Abstract
Rationale: Longitudinal data on the impact of continued, switched or discontinued antifibrotic therapy in patients with idiopathic pulmonary fibrosis (IPF) who have disease progression is needed.
Objective: We hypothesized that ongoing antifibrotic use (versus discontinuation) in the setting of forced vital capacity (FVC) decline would be associated with less future decline and lower likelihood of a composite outcome of FVC decline, lung transplant, or death.
Methods: We performed a multicenter cohort study using data from the Canadian Registry for Pulmonary Fibrosis in patients with IPF with FVC decline ≥10% over 6 months on antifibrotic therapy. The association of continued, switched or discontinued therapy with (1) further change in FVC and (2) a composite of FVC decline ≥10%, transplant, or death, in the subsequent 6 months, was assessed using adjusted linear and logistic regression modelling, respectively. Generalized estimating equations accounted for repeated observations per patient.
Results: 165 patients had a decline in FVC ≥10% over 6 months while receiving antifibrotic therapy. Compared to continued use, antifibrotic discontinuation after FVC decline was associated with greater additional FVC decline (-207 mL 95%CI -353 to -62, p = 0.005) and higher odds of FVC decline ≥10%, transplant, or death (odds ratio 12.2 95%CI 1.2 to 130.5, p = 0.04). There was no difference between continued versus switched antifibrotic therapy.
Conclusions: Ongoing antifibrotic therapy in the setting of FVC decline is associated with less future FVC decline and lower odds of FVC decline ≥10%, transplant, or death in a real-world cohort of IPF.
Keywords: Antifibrotics; Idiopathic pulmonary fibrosis; Interstitial lung disease.
A cluster-based analysis evaluating the impact of comorbidities in fibrotic interstitial lung disease
Abstract
Background: Comorbidities are frequent and have been associated with poor quality of life, increased hospitalizations, and mortality in patients with interstitial lung disease (ILD). However, it is unclear how comorbidities lead to these negative outcomes and whether they could influence ILD disease progression. The goal of this study was to identify clusters of patients based on similar comorbidity profiles and to determine whether these clusters were associated with rate of lung function decline and/or mortality.
Methods: Patients with a major fibrotic ILD (idiopathic pulmonary fibrosis (IPF), fibrotic hypersensitivity pneumonitis, connective tissue disease-associated ILD, and unclassifiable ILD) from the CAnadian REgistry for Pulmonary Fibrosis (CARE-PF) were included. Hierarchical agglomerative clustering of comorbidities, age, sex, and smoking pack-years was conducted for each ILD subtype to identify combinations of these features that frequently occurred together in patients. The association between clusters and change in lung function over time was determined using linear mixed effects modeling, with adjustment for age, sex, and smoking pack-years. Kaplan Meier curves were used to assess differences in survival between the clusters.
Results: Discrete clusters were identified within each fibrotic ILD. In IPF, males with obstructive sleep apnea (OSA) had more rapid decline in FVC %-predicted (− 11.9% per year [95% CI − 15.3, − 8.5]) compared to females without any comorbidities (− 8.1% per year [95% CI − 13.6, − 2.7]; p = 0.03). Females without comorbidities also had significantly longer survival compared to all other IPF clusters. There were no significant differences in rate of lung function decline or survival between clusters in the other fibrotic ILD subtypes.
Conclusions: The combination of male sex and OSA may portend worse outcomes in IPF. Further research is required to elucidate the interplay between sex and comorbidities in ILD, as well as the role of OSA in ILD disease progression.
Costs of Workplace Productivity Loss in Patients with Connective Tissue Disease–associated Interstitial Lung Disease
Abstract
Rationale: Interstitial lung disease (ILD) develops in a large percentage of patients with connective tissue disease (CTD) and is associated with increased morbidity and mortality. Patients with CTD-associated ILD (CTD-ILD) often present at a young age, suggesting that ILD likely impacts workplace productivity.
Objectives: We aimed to determine the employment rate and workplace productivity loss, along with its associated factors and estimated costs, in patients with fibrotic CTD-ILD.
Methods: Patients with fibrotic CTD-ILD from the six centers of the Canadian Registry for Pulmonary Fibrosis were eligible. Health-related productivity loss was assessed using the Work Productivity and Activity Impairment questionnaire. Proposed factors associated with low workplace productivity were forced into a multivariable regression model. Average productivity loss in hours/week was used to calculate the costs of productivity loss based on hourly wages obtained from national census data matched for age and sex. Workplace productivity loss outcomes were compared between patients with CTD-ILD and patients with a non-CTD fibrotic ILD.
Results: Of 375 eligible patients with fibrotic CTD-ILD, 113 (30%) were employed. Productivity loss was reported by 59% of employed patients, with a mean loss of 9.4 ± 1.2 hours/week, including 3.9 ± 0.9 hours/week from absenteeism and 5.5 ± 0.7 hours/week from presenteeism. Employment among patients 25–54 years of age with fibrotic CTD-ILD was 27% lower than that in the matched general Canadian population (56% vs. 83%; P < 0.001). Employment among patients ≥55 years of age with CTD-ILD was 17% lower than that in the matched population (19% vs. 36%; P < 0.001). Workplace productivity loss was not associated with respiratory symptoms or lung physiology. Annual costs of productivity loss were calculated at 13,593 Canadian dollars per employee with fibrotic CTD-ILD. Workplace productivity loss was similar in patients with fibrotic CTD-ILD and those with non-CTD fibrotic ILD.
Conclusions: Patients with fibrotic CTD-ILD frequently report workplace productivity loss, which is unexplained by respiratory symptoms or lung physiology and is associated with significant costs.
Keywords: connective tissue disease; interstitial lung disease; workplace productivity
Treatment of rheumatoid arthritis-associated interstitial lung disease in a multi-center registry cohort
Abstract
Background: Rheumatoid arthritis-associated interstitial lung disease (RA-ILD) is challenging to manage, with a paucity of robust data to guide treatment. Our aim was to characterize the pharmacologic treatment of RA-ILD utilizing a retrospective design in a national multi-center prospective cohort, and to identify associations between treatment and change in lung function and survival.
Methods: Patients with RA-ILD and a radiological pattern of non-specific interstitial pneumonia (NSIP) or usual interstitial pneumonia (UIP) were included. Unadjusted and adjusted linear mixed models and Cox proportional hazards models were used to compare lung function change and risk of death or lung transplant by radiologic patterns and treatment.
Results: Of 161 patients with RA-ILD, UIP pattern was more common than NSIP (55.9% vs. 44.1%). Only 44/161 (27%) patients were treated over median follow-up of 4 years with medication choice appearing unrelated to patient-specific variables. Decline in forced vital capacity (FVC) was not associated with treatment. Patients with NSIP had lower risk of death or transplant, compared to UIP (P=0.0042). In patients with NSIP, there was no difference in time to death or transplant comparing treated to untreated in adjusted models [hazard ratio (HR) =0.73; 95% confidence interval (CI): 0.15–3.62; P=0.70]. Similarly, in patients with UIP, there was no difference in time to death or lung transplant between treated and untreated in adjusted models (HR =1.06; 95% CI: 0.49–2.28; P=0.89).
Conclusions: Treatment of RA-ILD is heterogeneous, with most patients in this cohort not receiving treatment. Patients with UIP had worse outcomes compared to NSIP, similar to other cohorts. Randomized clinical trials are needed to inform pharmacologic therapy in this patient population.
Keywords: Connective tissue disease, diffuse parenchymal lung disease, pulmonary fibrosis, lung function, outcomes
Data Collection Sites
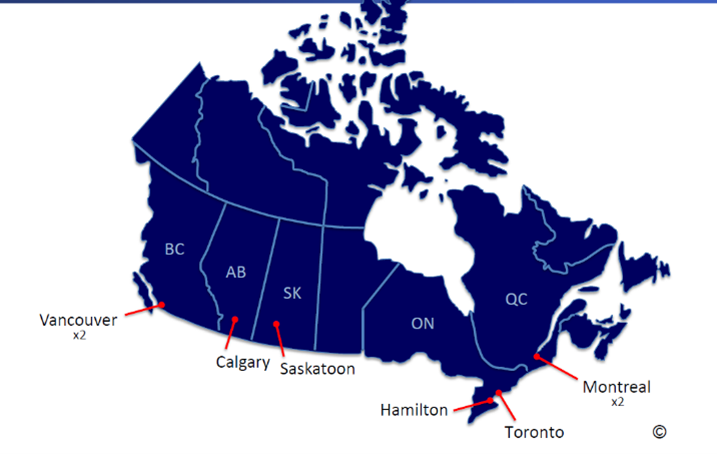
CARE-PF gathers valuable data through eight collection sites across Vancouver, Calgary, Saskatoon, Hamilton, Toronto, and Montreal. Below are the participating institutions and their points of contact:
- McGill University
- Contact: Deborah Assayag
- Email: deborah.assayag@mail.mcgill.ca
- McMaster University
- Contact: Victoria
- Email: bryczkav@mcmaster.ca
- University of British Columbia (St. Paul’s Hospital)
- Contact: Likhitha Marni
- Email: lmarni@btrg.ca
- University of British Columbia (Vancouver General Hospital)
- Contact: Marion Santos
- Email: marion.santos@vch.ca
- University of Calgary
- Contact: Rebecca Bruke
- Email: rebecca.burke@ucalgary.ca
- University of Montreal
- Contact: Celine Durand
- Email: celine.durand.chum@ssss.gouv.qc.ca
- University of Saskatchewan
- Contact: Veronica Marcoux
- Email: vsm940@mail.usask.ca
- University of Toronto
- Contact: Natana Keji
- Email: keji.natana@uhn.ca
For site-specific inquiries, please directly connect with site contacts. To propose a new site, use our contact form to submit your inquiry.
Data Security
Our team takes data security seriously and ensures that data collected at our sites is stored on University of British Columbia’s secure server located at St. Paul’s Hospital. The database is safeguarded through robust security measures, including:
- Password Encryption: Access to the database requires the use of encrypted passwords.
- Dual-Factor Authentication: Additional security is ensured through dual-factor authentication for authorized users.
- Institutional Firewalls: The server is protected by institutional firewalls to prevent unauthorized access.
Access to Data:
Access to the database is strictly controlled and limited to authorized personnel. Approval from the Principal Investigator is required for any access to the database.
Data Anonymization:
Patient information is anonymized by assigning a unique study number to each participant. This number does not include any personal identifying information, ensuring participant confidentiality.
If you have any questions about data security, please fill out our contact form with your inquiry: